Scientific Achievement
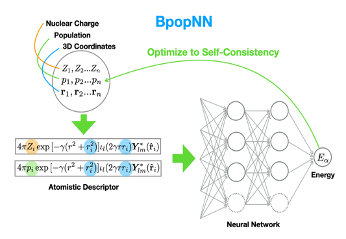
DFT is a primary tool for studying the electronic properties of materials. DFT’s description can be simplified to be based on elementary electronic features of the material’s atoms. We use ML to model this description. This is a novel way to incorporate electronic information, e.g. polarization, into ML potential energy surface (PES) approximations.
Significance and Impact
Electronic properties have been largely ignored in ML molecular models, which is problematic for studying charged species and redox reactions abundant in battery environments. Our model provides a route for solving this problem. This will open up possibilities for large scale molecular simulations for both charged/uncharged species at DFT accuracy with much reduced computational cost.
Research Details
For the elementary electronic features, we used atomic electron populations, the number of electrons belonging to a particular atom.
Our ML model, called BpopNN, allows the electronic features to adapt to each other in a consistent and molecule-specific way.
We confirmed the effectiveness of this approach with a variety of calculations on LinHn clusters.
