Scientific Achievement
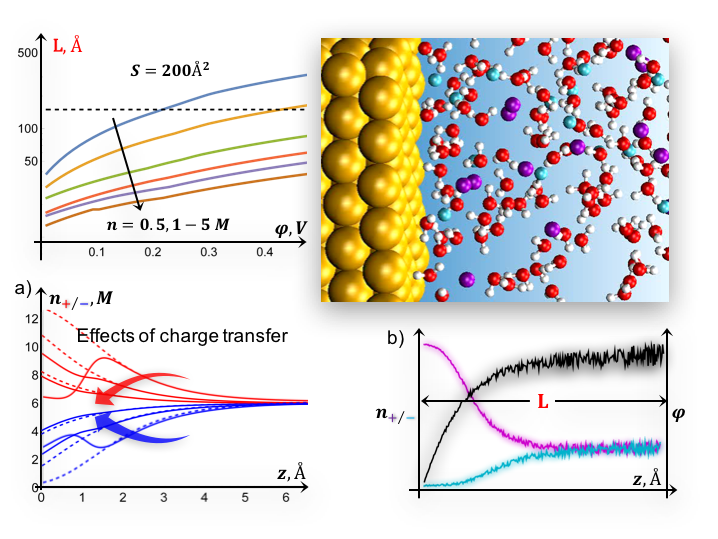
We develop a self-consistent methodology for modeling biased interfaces that combines (i) continuum theory and (ii) ab initio molecular dynamics, to explore the structure of the electric double layer under various electrochemical conditions, including effects of electron transfer and non-electrostatic interactions (e.g. adsorption).
Significance and Impact
Ab initio molecular dynamics studies aiming to describe biased interfaces are often computationally limited to small sizes, suffer from inconsistency with boundary conditions (concentration, surface charge, etc.) and large fluctuations that may compromise stability. Here we provide estimates of minimum requirements for a microscopic description in terms of the cell sizes of ab initio molecular dynamics setups and the applied surface charge density/electrode potential.
Research Details
- The potential-dependent charge distribution in the double layer close to redox levels can be efficiently performed with a formalism based on a pair of coupled modified Poisson-Boltzmann equations
- For realistic electrolyte concentrations (~1M) and voltages (~0.1V), reliable AIMD simulations can be designed based on a continuum model, avoiding strongly non-equilibrium configurations and incompatibility with the target external conditions
