Scientific Achievement
- Molecular details of Mg ion solvation in a proven electrolyte explored with ab initio molecular dynamics
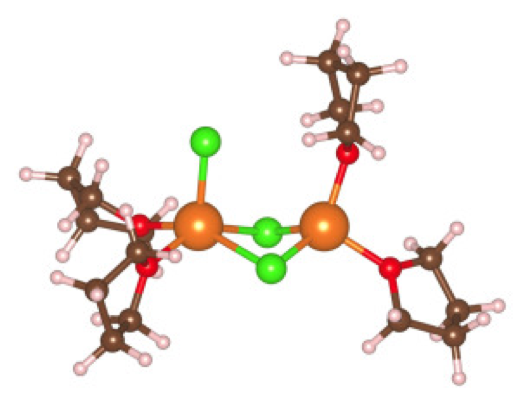
- Mg ions remain bound to Cl anions and are typically 4-fold coordinated at finite temperature in solution. Mg dimer complexes can be at most 5-fold coordinated and exhibit solution phase structures in stark contrast to crystals
- Simulated X-ray absorption spectra indicate clear differences between 6- and 4-fold coordination
Significance and Impact
- Need for first-principles modeling – existing parametrized force fields predict incorrect coordination
- Mg ion diffusion and extraction should be enhanced with lower coordination
- Performance bottlenecks most likely defined by interfacial effects at electrode-electrolyte interface
- Future X-ray characterization can confidently focus clear spectral distinctions in Mg coordination near electrodes
Research Details
- First-principles molecular dynamics simulations were performed using high-performance computing resources at NERSC
- X-ray absorption calculations use an established approach developed at the Molecular Foundry, providing significant motivation for ALS experiments
Work performed at The Molecular Foundry, Lawrence Berkeley National Laboratory (JCESR partner) by L. F. Wan and D. Prendergast, Journal of the American Chemical Society, 2014.
DOI: 10.1021/ja505967u
