Scientific Achievement
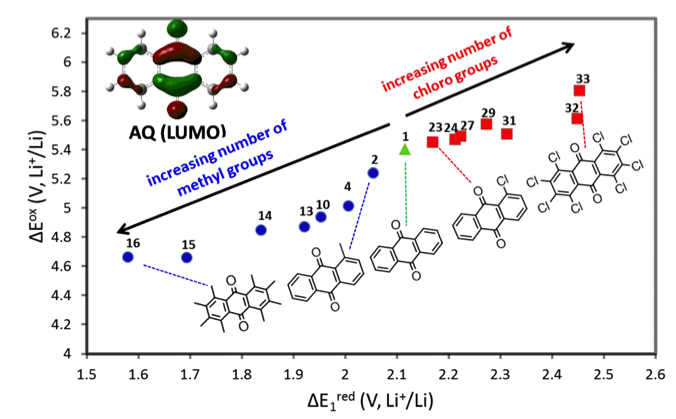
Simulations provided the contribution of functional groups toward better electrochemical properties and solubility of Anthraquinone (AQ) derivatives, a promising class of compounds for aqueous and non-aqueous redox flow applications.
Significance and Impact
The discovery of these novel derivatives opens up a new horizon of high performance redox active organic materials for storage applications.
Research Details
- Computational studies were used to predict electrochemical windows of ~50 Anthraquinone derivatives.
- Computations indicate that complete methylation of AQ can improve its reduction window by ∼0.4 V.
- Derived a relationship that connects the computed LUMO energy and the reduction potential that can be applied as a descriptor for screening thousands of AQ derivatives.
- Our computations suggest that incorporating oxy-methyl dioxolane substituents in the AQ framework may increase its solubility.
Work performed at Argonne National Laboratory, (JCESR managing partner) by J. E. Bachman, L. A. Curtiss, R. S. Assary, Investigation of the Redox Chemistry of Anthraquinone Derivatives Using Density functional Theory, J. Phys. Chem. A, 2014, 118, 8852-8860.
DOI: 10.1021/jp5060777
