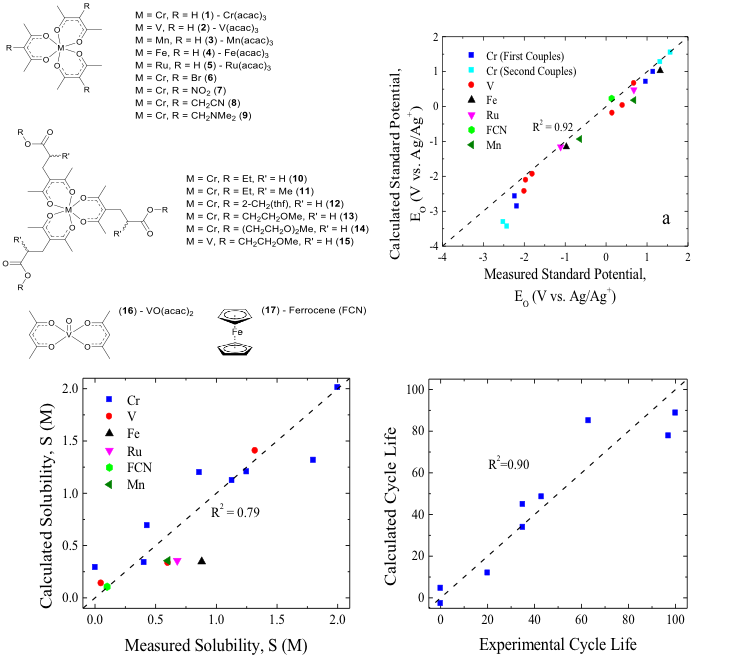
Scientific Achievement
Density functional theory (DFT) was used to calculate key materials properties that were correlated with experimentally determined parameters that define the performance of redox flow battery (RFB) active materials. These include standard potentials, solubilities, and importantly stabilities. The correlations are for metal-acetylacetonate (acac) complexes, a promising class of actives, but could also be used for other materials.
Significance and Impact
The structure-function relationships and correlations presented in this paper could be used to predict new, highly soluble and stable complexes, thus enabling the synthesis of better RFB chemistries.
Research Details
- The modeled standard potentials were in very good agreement with the experimental results for a variety of acac complexes.
- The solubility correlation was generated using the solvation energy and the dipole moment as descriptors; the result can be used to screen for complexes with high solubilities within the same class (i.e. acacs), thus reducing the experimental burden.
- Cycle lifes determined using bulk electrolysis correlated linearly with the percent of the LUMO/HOMO on the metal center determined using DFT. This indicates that primarily metal-based redox reactions are more stable than reactions that involve significant charge storage on the ligand. The derived relationships could be used to predict the cycle life of redox couples for new complexes of this type.
